Koch's
postulates state certain conditions
which must be fulfilled in order to demonstrate the etiology of infectious
disease. These postulates are:
The
microorganism must regularly be isolated from cases of the disease.
It
must be grown in pure culture in vitro.
When
such a pure culture is inoculated into susceptible animal species, the
typical disease must result.
The
microorganism must be re-isolated from the susceptible animal.
Prions
Structure
Prions
are infectious agents composed exclusively of a single sialoglycoprotein
called PrP 27-30. They contain no nucleic acid. PrP 27-30 has a mass of
27,000 - 30,000 daltons and is composed of 145 amino acids with glycosylation
at or near amino acids 181 and 197. The carboxy terminus contains a phosphatidylinositol
glycolipid whose components are ethanolamine, phosphate, myo-inositol and
stearic acid. This protein polymerizes into rods possessing the ultrastructural
and histochemical characteristics of amyloid. Amyloid
is a generic term referring to any optically homogenous, waxy, translucent
glycoprotein; it is deposited intercellularly and/or intracellularly in
many human diseases such as:
Alzheimer's
disease
Creutzfeldt-Jakob
disease
Down's
syndrome
Fatal
familial insomnia
Gerstmann-Straussler
syndrome
Kuru
Leprosy
Replication
The
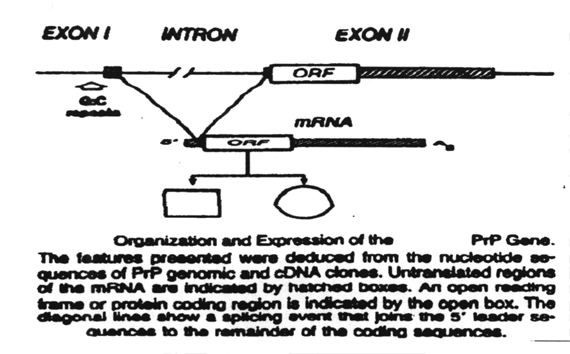
prion is a product of a human gene, termed the PrP gene, found on chromosome
20. This gene contains two exons separated by a single intron. Exon I and
Exon II are transcribed and the two RNAs ligated into a single mRNA. This
mRNA contains an open reading frame (ORF) or protein coding region which
is translated into the PrP protein. The PrP protein is a precursor of the
prion protein. It is termed PrP 33-35.
The
PrP 33-35 undergoes several post-translational events to become the prion
protein (PrP 27-30):
1.
Glycosylation - at two sites.
2.
Formation of a disulfide bond between two cysteine residues.
3.
Removal of the N-terminal signal peptide.
4.
Removal of the C-terminal hydrophobic segment.
5.
Addition of a phosphatidylinositol glycolipid at the C-terminal.
6.
Removal of the N-terminal first 57 amino acids.
In
normal cells only the PrP 33-35 protein is synthesized. It is found in
the neural cell membrane where it's function is to sequester Cu++ ions.
In abnormal ("infected") cells, the PrP 27-30 is produced from the PrP
33-35 protein. The PrP 27-30 triggers a series of reactions that produce
more PrP 27-30 proteins, i.e., PrP 27-30 induces its own synthesis. In
addition to the post translational modifications, the PrP 27-30 protein
differs from the PrP 33-35 protein in a single amino acid residue. Residue
178 in the PrP 27-30 contains an asparagine residue whereas the PrP 33-35
protein has an aspartate residue at this position. This causes a conformational
change in the PrP 27-30 protein from an -helix to a -sheet. This conformational
change in the PrP 27-30 protein has three effects:
1.
It imparts to the PrP 27-30 protein the ability to induce the same -helix
to -sheet conformation in the PrP 33-35 protein. This is a permanent conformational
change. It thus induces its own "replication."
2.
The -sheet-forming peptides aggregate to form amyloid fibrils.
3.
The amyloid fibrils kill thalamus neurons through apoptosis, a programmed
series of events that leads to cell death.
Pathologies
induced by prions
All
diseases known to be of prion etiology, in animals and humans, are neurodegenerative
diseases. In the human this includes:
Creutzfeldt-Jakob
disease (CJD)
Fatal
Familial Insomnia
Gerstmann-Straussler
syndrome
Kuru
The
pathological and clinical signs of these diseases suggest that they are
closely related. In fact they may be variants of the same disorder. All
pathological features are confined to the central nervous system. The prion
protein accumulates selectively and abnormally in CNS nerve cells during
the course of the disease. PrP 27-30 accumulates within the neuropil where
it causes:
1.
Astrocyte gliosis (an increase in the number of astrocytes).
2.
Depletion of dendritic spines in neurons.
3.
Formation of numerous vacuoles in the cerebellar cortex (spongiform
encephalopathy).
4.
Amyloidosis - deposition of amyloid in the cerebellar cortex, thalamus,
brain stem and in the lumen of blood vessels within the brain. These amyloid
plaques consist of discrete eosinophilic glassy-appearing masses, often
having radiating amyloid fibrils at their periphery. The plaques are primarily
subependymal, subpial and perivascular.
Note
that the pathology does NOT include any signs of inflammation
or fever. This is evidence that the immune system does not respond to the
prion protein. Since the prion protein is derived from self this is what
you would expect.
These
pathologies give rise to the clinical symptomology seen in these patients.
These are:
1.
A long incubation period (several years) which has given rise to the term
"slow infection."
2.
Loss of muscle coordination which leads to a difficulty in walking, indicating
a functional disorder of the cerebellum.
3.
Dementia characterized initially by loss of memory, diminished intellect
and poor judgement.
4.
Progressive insomnia characterized by a marked reduction or loss of the
slow-wave and rapid-eye-movement phases. D. Transmission
Spread
of the disease is via horizontal
transmission, i.e., transmission from
one person to another, either directly or by fomites.
Viroids
Structure
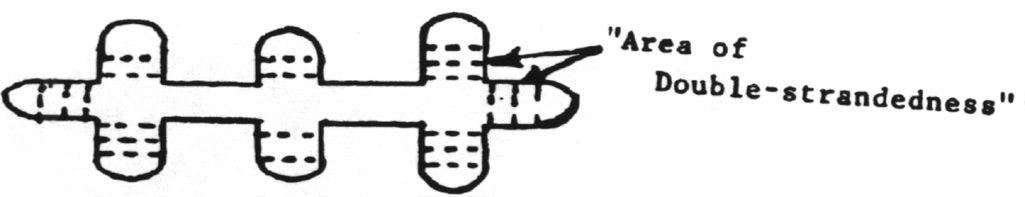
Viroids
are infectious agents composed exclusively of a single piece of circular
single stranded RNA which has some double-stranded regions.
Because
of their simplified structures both prions and viroids are sometimes called subviral
particles. Viroids mainly cause plant
diseases but have recently been reported to cause a human disease.
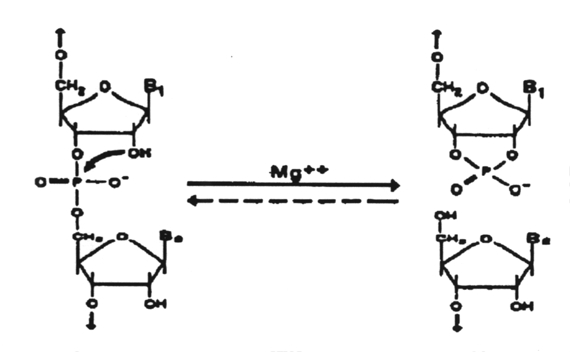
Catalytic
RNAs are those that have the intrinsic ability to break and form covalent
bonds; Viroids are catalytic RNA's (ribozymes) that cleave RNA to produce
fragments containing a 5'-hydroxyl and a 2', 3'-cyclic phosphate.
This
is a nonhydrolytic reaction in which the same number of phosphodiester
bonds are maintained and the transesterification reaction is theoretically
reversible. This reaction is considered to play an essential role in the
replication of these RNAs in vivo. Such reactions are all intramolecular
and hence quasi-catalytic with single turnover. These RNAs can be manipulated,
however, to provide true catalytic cleavage in trans-reactions.
Replication
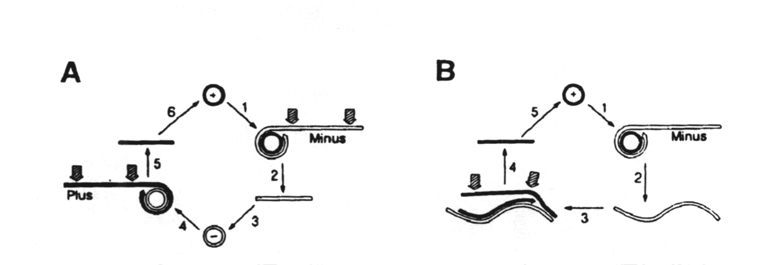
Circular,
pathogenic RNAs are replicated by a rolling circle mechanism in vivo.
There are two variations of this rolling circle mechanism:
In
the first variation (A), the circular plus strand is copied by viroid RNA-dependent
RNA polymerase to form a concatameric minus strand (step 2). Site-specific
cleavage (arrows) of this strand produces a monomer that is circularized
by a host RNA ligase (step 3) and then copied by the RNA polymerase to
produce a concatameric plus strand. Cleavage of this strand (step 5) produces
monomers which, on circularization, produces the progeny circular, plus
RNA, the dominant form in vivo.
In
the other variation (B), the concatameric minus strand of step 1 is not
cleaved but is copied directly to give a concatameric plus strand (step
3), which is cleared specifically to monomers for ligation to the circular
progeny. Those RNAs that self-cleave only in the plus strand in vitro
are considered to follow this route.
Human
pathologies induced by viroids
The
only human disease known to be caused by a viroid is hepatitis D. This
disease was previously ascribed to a defective virus called the delta
agent. However, it now is known that
the delta agent is a viroid enclosed in a hepatitis B virus capsid. For
hepatitis D to occur there must be simultaneous infection of a cell with
both the hepatitis B virus and the hepatitis D viroid. There is extensive
sequence complementarity between the hepatitis D viroid RNA and human liver
cell 7S RNA, a small cytoplasmic RNA that is a component of the signal
recognition particle, the structure involved in the translocation of secretory
and membrane-associated particles. The hepatitis D viroid causes liver
cell death via sequestering this 7S RNA and/or cleaving it.
Transmission
The
hepatitis D viroid can only enter a human liver cell if it is enclosed
in a capsid that contains a binding protein. It obtains this from the hepatitis
B virus. The delta agent then enters the blood stream and can be transmitted
via blood or serum transfusions.
References
Prusiner,
S.B., 1991. Molecular Biology of Prion Diseases. Science 252:1515-1522.
Medori,
R., et al., 1992. Fatal Familial Insomnia, A Prion Disease with
a Mutation at Codon 178 of the Prion Protein Gene. The New England Journal
of Medicine 326:444-449.
Taylor,
J.M., 1992. The Structure and Replication of Hepatitis Delta Virus. Annual
Reviews of Microbiology 46:253-276.
Touchette,
N., 1993. ß-sheet structure is key issue in prion disease. The Journal
of NIH Research 5:57-59.
Barinaga,
M., 1993. Ribozymes: killing the messenger. Science 263:1512-1514.
Prusiner,
S.B., 1995. The Prion Diseases. Scientific American 272: 48-57.
Summary
1.
Koch's postulates are a means of relating a given set of clinical symptoms
to infection with a particular etiological agent.
2.
Prions are infectious agents composed solely of glycoprotein. They are
products of a human gene which accumulate in tissue as amyloid.
3.
Amyloid deposition in tissue is a pathological manifestation of many diseases,
of both prion and non-prion etiology. These diseases include Alzheimer's
disease, *Creutzfeldt-Jakob disease, Down's syndrome (mongolism), *fatal
familial insomnia, *Gerstmann-Straussler syndrome, *kuru and leprosy. (*
indicates prion diseases).
4.
The accumulation of amyloid induces these pathologies in the host: astrocyte
gliosis, depletion of neuronal dendritic spines, spongiform encephalopathy.
5.
In prion disease there is a long incubation period before one sees loss
of muscle coordination, dementia and/or progressive insomnia.
6.
Prions induce no immune reactions within the human.
7.
Viroids are infectious agents composed solely of circular single-stranded
RNA which folds over on itself to form some double stranded regions. These
are catalytic RNAs (ribozymes).
8.
The only human disease known to be caused by a viroid is hepatitis D; in
this case the viroid is enclosed in a hepatitis B virus capsule.
9.
The hepatitis D viroid manifests its disease potential by sequestering
and/or destroying human liver 7S RNA.